





视网膜色素变性(RP)是一种导致视力丧失的眼睛遗传性疾病。[1]症状包括夜间观察困难和周边视力下降(侧视力)。[1]症状的发作通常是渐进的。[2]随着周边视力的恶化,人们可能会体验到“隧道视觉”。[1]完全失明并不常见。[2]
色素性视网膜炎通常遗传自一个人的父母。[1]涉及超过50个基因之一的突变。[1]潜在的机制涉及眼后部的视杆细胞逐渐丧失。[1]这通常会导致锥形感光细胞的丢失。[1]诊断是通过检查视网膜发现暗色素沉积物。[1]其他支持性测试可能包括视网膜电图,视野测试或基因测试。[1]
目前尚无治疗视网膜色素变性的方法。[2]管理问题的努力可能包括使用低视力辅助设备,便携式照明设备或导盲犬。[1]维生素A棕榈酸酯补充剂可能有助于减缓恶化。[1]视觉假体可能是某些患有严重疾病的人的选择。[1]估计会影响到4,000人中的1人。[1]发病通常发生在儿童时期,但有些人直到成年才受到影响。[1] [2]
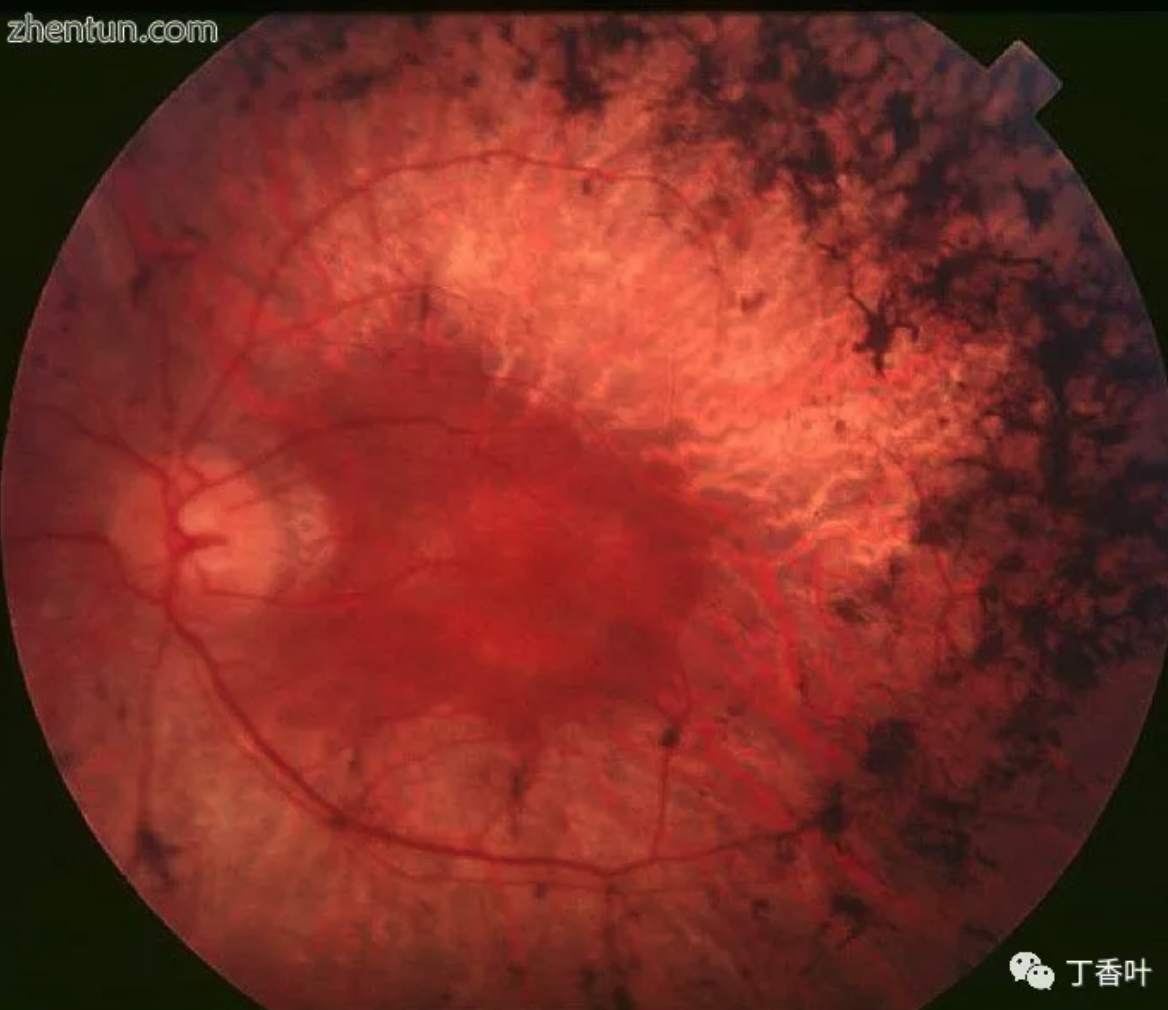
背部视网膜色素变性患者的眼睛,中期。 注意中间周围的色素沉积以及视网膜萎缩。 虽然保留黄斑,但周围有一些色素沉着损失。
目录
1 症状和体征
2 原因
2.1 遗传学
3 病理生理学
4 诊断
5 治疗
6 预后
7 流行病学
8 研究
9 值得注意的案例
10 参考资料
体征和症状

隧道视觉示例(下)
视网膜色素变性的初始视网膜退行性症状的特征是夜视(夜盲症)减少和中周视野丧失[3]。杆感光细胞负责低光视力并在视网膜周边定向,是在这种疾病的非综合征形式期间首先受影响的视网膜过程。[4]视觉下降相对较快地进入远边缘区域,最终在隧道视觉增加时延伸到中心视野。由于锥形感光细胞伴随异常,视敏度和色觉可能会受到损害,这些细胞负责中央视野中的色觉,视敏度和视力。[4]疾病症状的进展以对称的方式发生,左眼和右眼都以相似的速率出现症状。[5]
各种间接症状表征视网膜色素变性以及初始视杆受体变性和后来视锥感光器衰退的直接影响。诸如畏光的现象,其描述了光被感知为强烈眩光的事件,以及视野中存在闪烁或闪烁的光,在RP的后期阶段经常出现。与RP相关的发现通常在眼底被描述为“眼科三联症”。这包括(1)由骨针形成引起的视网膜色素上皮(RPE)的斑驳外观,(2)视神经的蜡状外观,和(3)视网膜中血管的出现。[ 3]
非综合征RP通常会出现以下各种症状:
夜盲症
隧道视力(由于周边视力丧失)
格子视觉
光幻视(闪烁/闪烁灯)
畏光(厌恶强光)
眼底骨针的发育
从暗到亮的环境缓慢调整,反之亦然
视力模糊
分色差
丧失中心视力
最终失明
原因RP可能是:(1)非综合征,即单独发生,没有任何其他临床发现,(2)综合症,其他神经感觉障碍,发育异常或复杂的临床表现,或(3)继发于其他系统疾病。[6]
RP与耳聋(先天性或进行性)相结合称为Usher综合征[7]。
Alport综合征与RP和肾小球基底膜异常相关,导致肾病综合征并继承为X连锁显性遗传。
在线粒体DNA疾病Kearns-Sayre综合征(也称为Ragged Red Fibre Myopathy)中可见RP伴有眼肌麻痹,吞咽困难,共济失调和心脏传导缺陷
在无β脂蛋白血症中可见RP与迟钝,周围神经病变,癫痫(加标)红细胞,共济失调,脂肪泻和无VLDL [8]相关。
作为McLeod综合征的一部分,RP临床上与其他几种罕见的遗传性疾病(包括肌营养不良症和慢性肉芽肿性疾病)相关。这是一种X连锁的隐性表型,其特征在于完全缺乏XK细胞表面蛋白,因此显着降低了所有Kell红细胞抗原的表达。对于输血目的,这些患者被认为与所有正常和K0 / K0供体完全不相容。
Bardet-Biedl综合征[9]可见与性腺机能减退有关的RP,以及伴有常染色体隐性遗传模式的发育迟缓
其他病症包括神经梅毒,弓形虫病和Refsum病。
遗传学色素性视网膜炎(RP)是遗传性视网膜变性最常见的形式之一[5]。
有多种基因在突变时可引起视网膜色素变性表型。[10] RP的遗传模式已被确定为常染色体显性遗传,常染色体隐性遗传,X连锁和母系(线粒体)获得,并且依赖于亲代中存在的特定RP基因突变。[11] 1989年,确定了视紫红质基因的突变,视紫红质是一种在视觉转导级联中发挥重要作用的色素,能够在弱光条件下实现视觉。视紫红质基因编码光感受器外部区段的主要蛋白质。该基因中的突变最常表现为视紫红质蛋白的错义突变或错误折叠,并且最常见于常染色体显性遗传模式。自发现视紫红质基因以来,已发现超过100种RHO突变,占所有类型视网膜变性的15%,以及约25%的常染色体显性形式的RP。[5] [12]
迄今为止,在与RP相关的视蛋白基因中已报道多达150个突变,因为该蛋白的盘内结构域中的Pro23 His突变在1990年首次报道。这些突变在整个视蛋白基因中发现并沿三个结构域分布。蛋白质(椎间盘内,跨膜区和细胞质区)。在视紫红质突变的情况下RP的主要生化原因之一是蛋白质错误折叠和分子伴侣的破坏[13]。发现视紫红质基因中密码子23的突变(其中脯氨酸变为组氨酸)占美国视紫红质突变的最大部分。其他几项研究报道了与视网膜色素变性有关的各种密码子突变,包括Thr58Arg,Pro347Leu,Pro347Ser,以及Ile-255的缺失。[12] [14] [15] [16] [17]在2000年,报道了密码子23的罕见突变导致常染色体显性视网膜色素变性,其中脯氨酸变为丙氨酸。然而,这项研究表明,与这种突变相关的视网膜营养不良在表现和病程方面具有特征性的轻微特征。此外,电子印迹幅度的保存比更普遍的Pro23His突变更多。[18]
已经在至少45个基因中鉴定了RP的常染色体隐性遗传模式。[11]这意味着两个未受影响的个体,其是双列型形式的相同RP诱导基因突变的携带者,可以产生具有RP表型的后代。已知USH2A基因的突变在以常染色体隐性遗传方式遗传时会引起10-15%的称为亚瑟氏综合征的综合征形式的RP [19]。
已知四种前mRNA剪接因子的突变导致常染色体显性遗传性视网膜色素变性。这些是PRPF3(人PRPF3是HPRPF3;也是PRP3),PRPF8,PRPF31和PAP1。这些因子无处不在地表达,并且提出普遍存在的因子(在任何地方表达的蛋白质)中的缺陷应该仅在视网膜中引起疾病​​,因为视网膜感光细胞对蛋白质加工(视紫红质)的需求远远高于任何其他细胞类型。 [20]
RP的体细胞或X连锁遗传模式目前用6个基因的突变鉴定,最常见的是发生在RPGR和RP2基因的特定位点。[19]
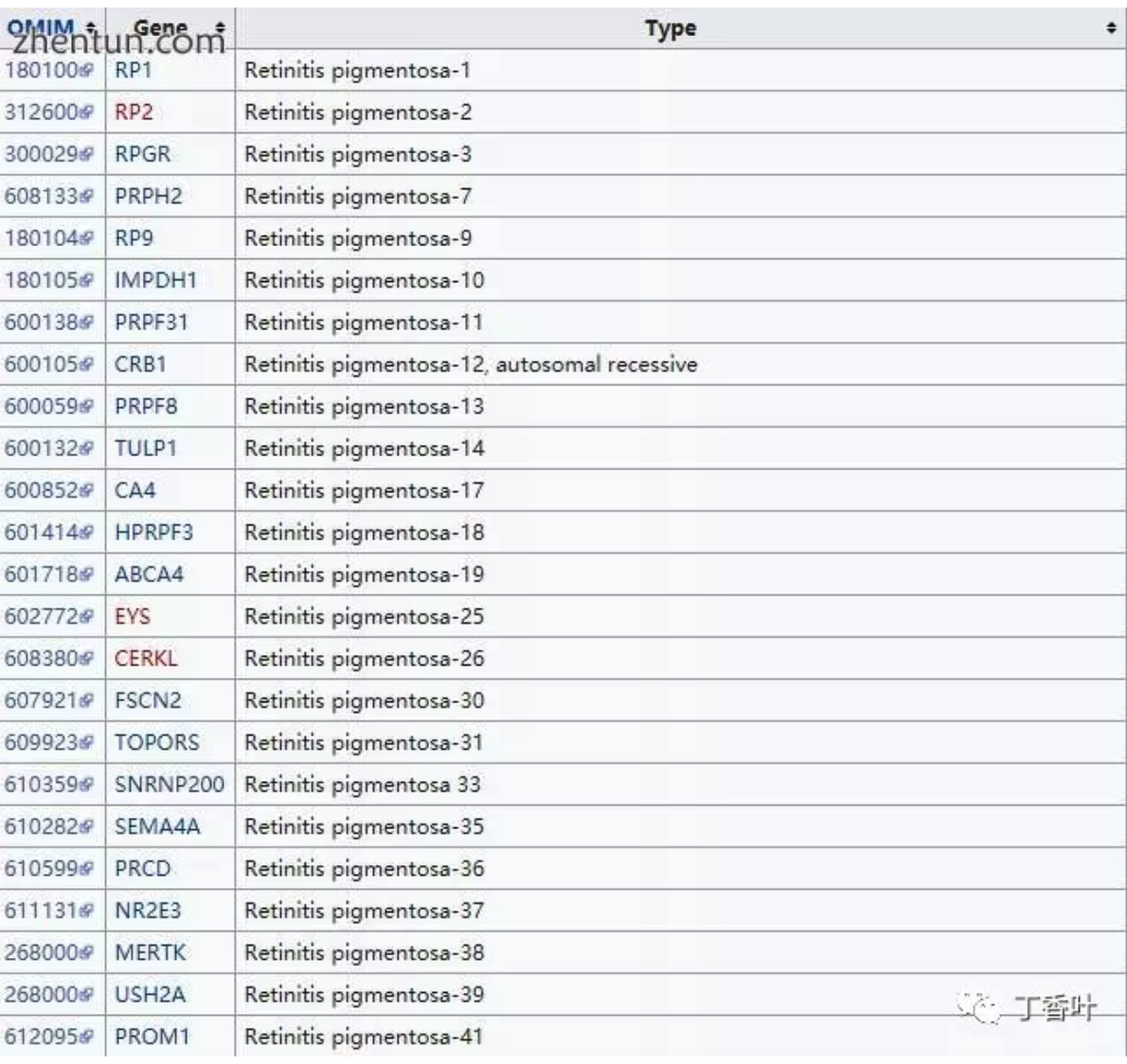
类型包括:
病理生理学

扫描电子显微镜照片描绘视网膜杆和视锥光感受器。细长杆呈黄色和橙色,而较短的锥体呈红色。
多种视网膜分子途径缺陷已与多种已知的RP基因突变相匹配。视紫红质基因的突变导致大多数常染色体显性遗传的RP病例,它破坏了在中枢神经系统的光转导级联中将光转化为可破译的电信号所必需的视杆蛋白。该G蛋白偶联受体活性的缺陷被分类为依赖于特定折叠异常和所得分子途径缺陷的不同类别。 I类突变蛋白的活性受到损害,因为蛋白质编码氨基酸序列中的特定点突变影响色素蛋白向眼外段的转运,其中光转导级联是局部的。此外,II类视紫红质基因突变的错误折叠破坏了蛋白质与11-顺式 - 视黄醛的结合,从而诱导适当的生色团形成。该色素编码基因中的其他突变体影响蛋白质稳定性,破坏翻译后mRNA的完整性,并影响转导蛋白和视蛋白光学蛋白的活化率。[21]
另外,动物模型表明视网膜色素上皮细胞不能吞噬已经脱落的外部杆区段盘,导致外部杆区段碎片的累积。在视网膜变性突变为纯合隐性的小鼠中,视杆细胞成熟完成前,视杆细胞感受器停止发育并发生变性。还记录了cGMP-磷酸二酯酶的缺陷;这导致cGMP的毒性水平。
诊断视网膜色素变性的准确诊断依赖于渐进性损失感光细胞功能的记录,通过视野和视力测试,眼底和光学相干图像以及视网膜电图(ERG)的组合证实,[22]
视野和视敏度测试通过与健康20/20视力相关的标准视觉测量来测量和比较患者视野的大小和视觉感知的清晰度。指示视网膜色素变性的临床诊断特征包括在视野测试中显着小且逐渐减小的视觉区域,以及在视敏度测试期间测量的受损透明度水平。[23]另外,在确定RP诊断时,诸如眼底和视网膜(光学相干)图像的光学断层摄影提供了进一步的诊断工具。拍摄扩张的眼睛的背部允许确认眼底中的骨针状物积聚,其在RP视网膜变性的后期阶段呈现。结合光学相干断层扫描的横截面图像,提供光感受器厚度,视网膜层形态和视网膜色素上皮生理学的线索,眼底图像可以帮助确定RP进展的状态。[24]
虽然视野和视力测试结果与视网膜图像相结合支持视网膜色素变性的诊断,但是需要额外的测试来确认该疾病的其他病理特征。视网膜电图(ERG)通过评估与光感受器变性相关的功能方面来确认RP诊断,并且可以在症状的初始表现之前检测生理异常。将电极透镜应用于眼睛,因为光感受器对不同程度的快速光脉冲的响应被测量。表现出视网膜色素变性表型的患者会在视杆光感受器中显示出降低或延迟的电响应,以及可能损害视锥细胞感光细胞的反应[25]。
由于视网膜色素变性的遗传模式,在确定诊断时也考虑患者的家族史。已知至少35种不同的基因或基因座会引起“非综合征RP”(RP不是另一种疾病或更广泛综合征的一部分)。 RP突变类型的适应症可通过DNA检测确定,该检测可在临床基础上获得:
RLBP1(常染色体隐性遗传,Bothnia型RP)
RP1(常染色体显性遗传,RP1)
RHO(常染色体显性遗传,RP4)
RDS(常染色体显性遗传,RP7)
PRPF8(常染色体显性遗传,RP13)
PRPF3(常染色体显性遗传,RP18)
CRB1(常染色体隐性遗传,RP12)
ABCA4(常染色体隐性遗传,RP19)
RPE65(常染色体隐性遗传,RP20)[26]
对于所有其他基因(例如DHDDS),分子遗传测试仅在研究基础上可用。
RP可以以常染色体显性遗传,常染色体隐性遗传或X连锁方式遗传。 X连锁RP可以是隐性的,主要影响雄性或显性,影响雄性和雌性,尽管雄性通常受到更轻微的影响。还描述了一些双基因(由两个基因控制)和线粒体形式。
遗传咨询取决于准确的诊断,确定每个家庭的遗传方式,以及分子遗传学检测的结果。
治疗目前尚无治愈视网膜色素变性的方法,但目前正在评估各种前瞻性治疗的疗效和安全性。各种补充剂(如维生素A,DHA和叶黄素)在延缓疾病进展方面的效率仍然是一个尚未解决的前瞻性治疗选择。[27] [28]研究视神经装置,基因治疗机制和视网膜片移植的临床试验是视网膜色素变性患者视力部分恢复的活跃研究领域。[29]
研究表明,每天摄入15000 IU(相当于4.5毫克)维生素A棕榈酸酯可延缓棒状光感受器变性;因此,阻碍了一些患者的疾病进展。[30]最近的研究表明,在疾病的某些阶段,适当的维生素A补充剂可以使失明延迟长达10年(通过将10%的损失降低至每年8.3%)。[31]
Argus视网膜假体于2011年2月成为该疾病的第一个批准治疗方案,目前可在德国,法国,意大利和英国使用。[32] 2012年公布了30例患者长期试验的中期结果。[33] Argus II视网膜植入物在美国也获得了市场认可。[34]该设备可以帮助已经失去感知形状和运动能力的RP的成年人更具移动性并进行日常活动。 2013年6月,美国的12家医院宣布他们将很快接受RP患者的咨询,为当年晚些时候Argus II的推出做准备。[35] [不可靠的医疗来源?] Alpha-IMS是一个视网膜下植入物,涉及手术植入视神经中央凹下方的小图像记录芯片。 Alpha-IMS研究的视觉改进措施要求在进行临床试验和获得市场批准之前证明设备的安全性。[36]
基因治疗研究的目标是以健康形式的基因病毒补充表达与视网膜色素变性表型相关的突变基因的视网膜细胞;因此,响应于与插入的健康基因相关的指令,允许视网膜感光细胞的修复和正常功能。研究健康RPE65基因插入视网膜中表达LCA2视网膜色素变性表型的临床试验测得视力适度改善;然而,视网膜光感受器的降解继续与疾病相关。[37]可能的是,基因治疗可以保留剩余的健康视网膜细胞,同时不能修复已经患病的感光细胞中早期的损伤累积。[29]对基因治疗的反应理论上会使表现出光感受器衰退进展最短的年轻患者受益;因此,通过健康的插入基因与更高的细胞拯救可能性相关联。[38]
预测色素性视网膜炎的进行性和缺乏明确治愈的原因导致该疾病患者的前景不可避免地令人沮丧。虽然完全失明是罕见的,[39]患者的视力和视野将继续下降,因为初始视杆感光器和后来的视锥光感受器退化进行。可能的治疗方法仍处于研究和临床试验阶段;然而,关于视网膜色素变性视觉恢复的治疗研究证明了未来的发展前景。
研究表明,携带疾病基因型的儿童可以从症状前咨询中获益,以便为渐进性视力丧失相关的身体和社会影响做好准备。虽然通过主动咨询可以稍微缓解心理预后[40],但疾病的物理意义和进展在很大程度上取决于初始症状表现的年龄和光感受器退化的速度,而不是获得前瞻性治疗。低视力专家提供的矫正视觉辅助和个性化视力治疗可以帮助患者纠正视力的轻微干扰并优化其剩余的视野。支持团体,视力保险和生活方式治疗是管理渐进性视力下降的其他有用工具。[22]
流行病学色素性视网膜炎是遗传性失明的主要原因,[41]大约有1/4,000个人在其一生中经历了非综合症状的疾病。[42]据估计,目前全世界有150万人受到影响。早发RP在生命的最初几年内发生,并且通常与症状疾病形式相关,而晚发性RP从早期到中期出现。
常染色体显性和隐性形式的视网膜色素变性同样影响男性和女性人群;然而,较不常见的X连锁形式影响X连锁突变的男性受体,而女性通常不受RP特性的影响。 X-连锁形式的疾病被认为是严重的,并且通常在后期阶段导致完全失明。在极少数情况下,X连锁基因突变的显性形式将同样影响男性和女性。[43]
由于RP的遗传遗传模式,许多分离群体表现出更高的疾病频率或特定RP突变的增加的流行。在视网膜色素变性中导致杆状光感受器变性的预先存在或正在出现的突变通过家族性传播;因此,允许某些RP病例集中到具有祖先疾病史的特定地理区域。已经进行了几项遗传研究,以确定缅因州(美国),伯明翰(英格兰),瑞士(影响1/7000),丹麦(影响1/2500)和挪威的不同流行率。[44]纳瓦霍印第安人也表现出RP遗传率的提高,估计影响了1878人中的1人。尽管特定家族系中RP的频率增加,但该疾病被认为是非歧视性的,并且往往同样影响所有世界人口。
研究未来的治疗可能涉及视网膜移植,人工视网膜植入,[45]基因治疗,干细胞,营养补充剂和/或药物治疗。
2006年:英国研究人员移植了处于发育高级阶段的小鼠干细胞,并且已经编程发育成感光细胞,进入基因诱导模拟人类视网膜色素变性和年龄相关性黄斑变性的小鼠。这些光感受器开发并与动物的视网膜神经细胞建立了必要的神经连接,这是恢复视力的关键步骤。以前认为成熟的视网膜没有再生能力。这项研究将来可能会导致人类使用移植来缓解失明。[46]
2008年:大阪生物科学研究所的科学家们发现了一种名为Pikachurin的蛋白质,他们认为这种蛋白质可以治疗色素性视网膜炎。[47] [48]
2008:色素性视网膜炎试图与FAM46A的基因表达相关联。[49]
2010年:一种可能的基因疗法似乎适用于小鼠。[50]
2012年:哥伦比亚大学医学中心的科学家们在动物模型上展示了基因治疗和诱导多能干细胞治疗可能是治疗视网膜色素变性的可行方案。[51] [52]
2012年:迈阿密大学巴斯康帕尔默眼科研究所的科学家们提供的数据显示,当眼睛注射了中脑星形胶质细胞源性神经营养因子(MANF)时,动物模型中的光感受器受到保护。[53] [54]
加利福尼亚大学伯克利分校的研究人员通过利用一种“光开关”来恢复盲目老鼠的视力,这种“光开关”可以激活受损的视杆细胞和视锥细胞的动物视网膜神经节细胞。[55]
2015年:Bakondi等人的一项研究。在Cedars-Sinai医疗中心发现,CRISPR / Cas9可用于治疗伴有视网膜色素变性的常染色体显性遗传形式的大鼠[56] [57]。
2016年:RetroSense Therapeutics旨在将来自光敏藻类的病毒注射到几个盲人(患有色素性视网膜炎)的人眼中。如果成功,他们将能够看到黑白。[58] [59]
2017年:FDA批准了一项名为Luxturna的基因疗法,用于治疗患有双等位基因RPE65突变的视网膜营养不良患者[60]。
值得注意的案例Neil Fachie,英国残奥会自行车手[61]
Lindy Hou,澳大利亚双人自行车运动员和铁人三项运动员[62]
Steve Lonegan,新泽西州波哥大市市长; 美国参议院共和党候选人[63]
Danelle Umstead,美国残奥会高山滑雪运动员,与星共舞(美国电视剧)选手[64]
Jon Wellner,美国演员[65]
Steve Wynn,美国商业巨头和拉斯维加斯赌场开发商[66]











